 Amplification
PCR
Amplification
PCR
 |
La réaction
PCR est réalisée sur 400 ng de plasmide pGFPuv, 240 ng de
chaque amorce et 7 unités de Taq DNA Polymerase dans un volume final
de 60 µl. 10µl de chaque réaction sont déposés
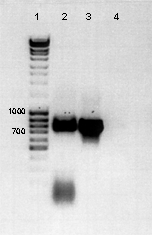
sur un gel de contrôle (Agarose 1% + BET 0.5µg/ml), ci-contre.
Piste1 : 4 µl Marqueur de
taille (MBI Fermentas). Piste 2 : réaction PCR réalisée
sur 400 ng de plasmide (sans huile minérale). Piste 3 : réaction
PCR réalisée sur 400ng de plasmide (avec huile minérale).
Piste 4 : réaction PCR réalisée sans plasmide.
Le contrôle négatif
en piste 4 certifie la spécificité de l'amplification PCR.
Le fragment d'ADN d'environ 800pb dans la piste 3 correspond à la
taille attendue pour le gène gfpuv. Les deux bandes observées
en piste 2, dont une correspond à la taille attendue et l'autre
beaucoup plus petite sont dues à un artefact réactionnel
expliqué dans "Recettes et astuces".
 Recettes
et astuces Recettes
et astuces |
Purification
de l'insert.
Le fragment issu de la PCR
(insert) est ensuite purifié en déposant le reste du volume
réactionnel sur un autre gel agarose 1% puis, après migration,
en découpant au scalpel la bande fluorescente correspondante. Le
morceau d'agarose contenant l'insert est placé dans un boudin de
dialyse pour y subir une électroélution. L'ADN en suspension
est alors précipité à froid dans de l'isopropanol,
resuspendu dans un plus petit volume et quantifié par spectrophotométrie
à 260nm.
Recettes
et astuces
Sous
clonage dans pGEMT
L'insert GFPuv ainsi purifié
est mélangé au plasmide pGEMT
ouvert dans un ratio (rapport) de 3:1, en présence d'ATP et de la
T4DNA Ligase pendant une heure
à température ambiante (au mois d'avril il n'est pas rare
que la température dépasse 25°C dans les salles de TP
!). Ces nouveaux plasmides sont alors introduits dans des bactéries
XL1blue (Escherichia coli) par électroporation. Les bactéries
effectivement transformées sont devenues résistantes à
un antibiotique de sélection du plasmide. Après une incubation
de 12 heures à 37°C, les différentes colonies résistantes
sont testées afin de vérifier lesquelles ont intégré
le vecteur pGEMT vide et lesquelles
ont intégré la nouvelle construction. Ce test est fait par
simple ouverture du plasmide et migration en gel d'agarose à 1%
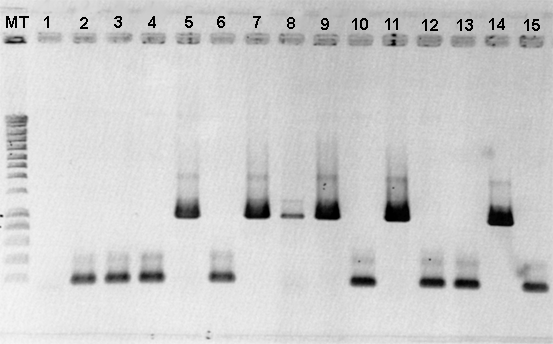
+ BET 0.5µg/ml. Les clones bactériens positifs (ceux recherchés),
possèdent un plasmide dont la taille est supérieure de 800pb
à celle du plasmide vide.
 |
Les clones bactériens ayant
proliféré sur milieu sélectif (c'est à dire
avec l'antibiotique) solide sont identifiés puis repiqués
sur milieu sélectif liquide et remis en culture. Ils subissent ensuite
une purification de leur ADN plasmidique après lyse bactérienne
suivie d'une précipitation (c'est la "miniprep").
Les préparations de plasmides
ainsi obtenues sont digérées pendant une heure par une endonucléase
ayant un seul site de clivage dans la construction vide ou "avec insert".
L'action d'une enzyme de restriction
facilite la lecture puisque le profils de migration electrophorétique
d'un plasmide est plus simple lorsque celui-ci est ouvert.
Les clones bactériens 5,
7, 9, 11 et 14 (ci-contre), contiennent un plasmide dont la taille correspond
à celle de pGEMT-GFPuv.
C'est à partir de ces clones
que nous poursuivrons l'étude.
Recettes
et astuces
|
Sous
clonage dans pGEX4T1.
Le gène gfpuv est
extrait du plasmide pGEMT-GFPuv
par action de l'endonucléase EcoRI. Le fragment ainsi découpé
est séparé du reste du plasmide au cours d'une migration
électophorétique en gel d'agarose 1%. L'insert est ensuite
électroélué et purifié.
En parallèle, le plasmide
pGEX4T1 est également
ouvert par EcoRI puis déphosphorylé par la phosphatase
alcaline. Cette étape de déphosphorylation est essentielle
lorsqu'une seule endonucléase est utilisée pour le sous clonage.
En effet, la probabilité que le plasmide se recircularise seul est
supérieure à celle qu'il a d'intégrer l'insert.
La phosphatase alcaline déphosphoryle les extrémités
5'-phosphate libres, or la T4DNA
Ligase a besoin de ce phosphate pour refermer définitivement les
plasmides. C'est ainsi que nous pouvons sélectionner les constructions
plasmidiques ayant intégré l'insert; elles sont les seules
a pouvoir être "recollées".
L'étape de ligation est
essentielle pour que le plasmide puisse être introduit durablement
dans une bactérie hôte.
Le gène gfpuvdigéré
par EcoRI et le plasmide pGEX4T1
ouvert et déphosphorylé sont mélangés avec
un ratio de 10:1 en présence d'ATP et de la T4DNA
Ligase pendant une heure à 37°C. La même réaction
est réalisée, en parallèle, avec le plasmide ouvert
déphosphorylé sans insert. Ce contrôle permet d'évaluer
l'efficacité de la déphosphorylation et d'en déduire
la probabilité d'insertion (si la déphosphorylation est efficace
à 100%, tous les clones bactériens capables de se multiplier
sur milieu sélectif ont intégré l'insert).
Les clones bactériens transformés
avec la construction pGEX4T1-GFPuv
peuvent être de deux types selon l'orientation de l'insertion : le
gène GFPuv peut être orienté dans le bon sens et donc
la traduction donnera la protéine de fusion, ou bien, le gène
est inséré dans le mauvais sens et la traduction en "verlen"
produira une protéine différente ... ou pas de protéine
du tout!
Cette discrimination est possible
en réalisant des minipreps des plasmides des clones bactériens
résistants puis en découpant ces plasmides avec BamHI.
Cette enzyme possède plusieurs sites de restriction sur pGEX4T1
et sur GFPuv et donc les profils de migrations seront différents
selon le sens d'orientation. Dans le cas où l'orientation est bonne,
des fragments d'ADN de 2 tailles différentes seront observés
: 544 et 5205pb. Si l'orientation est mauvaise, 3 fragment seront alors
observés : 201 544 et 5004pb.
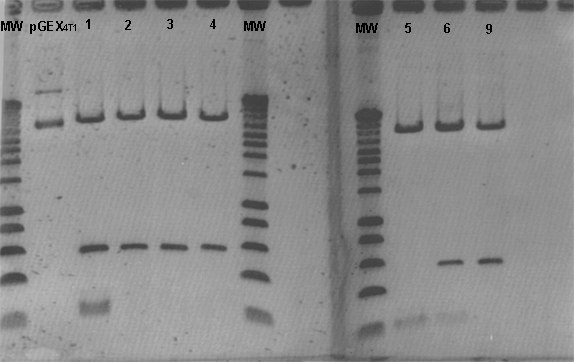 |
Les plasmides digérés
par BamHI sont séparés dans un gel d'agarose à 0,8%
(ci-contre).
Les clones 2, 3, 4 et 9 possèdent
le bon profil de migration, c'est à dire que les clones bactériens
correspondant ont intégré le plasmide avec la bonne orientaion,
tandis que les clones 1, 5 et 6 possèdent le profil de migration
carractéristique d'une mauvaise orientation.
Un des clones bactérien intéressant
sera utilisé pour la production et la caractérisation de
la protéine de fusion.
Recettes
et astuces
|
 5
5
[accueil] [principe
de la PCR] [recherche par thème]
[FAQ] [Glossaire]
[Liens spécifiques GFP] [Liens
externes]