
 Français
Français  English (UK)
English (UK) |
|
|
|
| Gene expression level in individual cells |
Cellular traits acquired on individual cells |
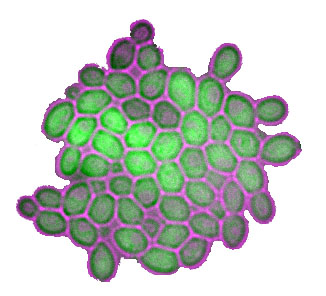



A human body is composed of many, many,... many cells. Estimating the exact number is not straightforward, but simple calculations can give a rough order of magnitude: 'about' 1014 cells. This is a big number. Yet, some physiological traits, such as cancer or developmental processes rely on what happened in very few cells, or even a single one. Modern biology therefore tries to apprehend a statistical description of the events that take place in individual cells.
We are investigating how the genetic background influences the probability laws of these single-cell events. From experiments that acquire gene expression levels or cellular morphology from thousands of individual cells, we investigate how the statistical properties of trait values are modified by genetic backgrounds. These probabilistic effects of genetic polymorphisms are highly relevant to the incomplete penetrance of genetic mutations (why a fraction of carriers do not express the associated phenotype).
Some time ago, we discovered that natural wild genetic backgrounds of S. cerevisiae can generate different levels of stochasticity in the expression of a model reporter gene. This stochasticity (high variance in a population of isogenic cells) is under polygenic control, and the molecular sources of this 'noise' can be identified by quantitative genetics mapping.
|
|
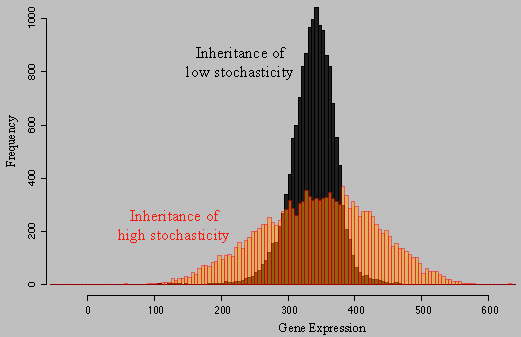
About the figure: Two independent wild genetic backgrounds of S. cerevisiae were crossed and segregants were analyzed for their level of a fluorescent protein in single cells. Each histogramm shows the distribution of MET17-GFP expression in isogenic cells of one segregant from this cross. In black, very low level of stochasticity was inherited, whereas the 'red' segregant inherited a very high level of stochasticity. We found that transcriptional elongation was partly involved in this difference. For details, please see our publications Ansel et al. 2008, Fehrmann et al. 2013, Yvert et al. 2013, Yvert 2014, Chuffart et al. 2016, Triqueneaux et al. 2020 |
We developed a novel statistical method that scans genomes for single-cell Probabilistic Trait Loci, which we called PTLMAPPER. For details, see Chuffart et al. 2016 here, and you can freely use our software, which is made available as an R package. See our download page for access.
We investigate these non-deterministic effects of natural genetic variations via Systems Biology approaches, which combine experimental acquisitions at high-throughput with mathematical modelling, predictions, experimental validations and deep DNA sequencing. Computer simulations benefit from the PSMN cluster resource of ENS de Lyon. For examples of such approaches and results see Richard et al. 2018 and Salignon et al. 2018 here.