
 Français
Français  English (UK)
English (UK) 





Responsable du thème: Gaël Yvert
|
|
|
|
| Expression d'un gène dans des cellules individuelles |
Caractères quantitatifs acquis sur des cellules uniques |
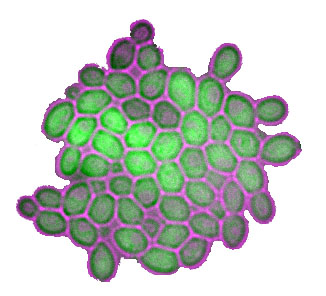



Le corps humain est composé d'un très, très, ... très grand nombre de cellules. En estimer le nombre exact est un exercice difficile, mais quelques calculs simples peuvent fournir un ordre de grandeur approximatif: 1014 cellules. C'est beaucoup. Or, certains caractères physiologiques, tels le cancer ou certaines étapes du développement, dépendent des évènements survenus dans très peu de cellules, voire une seule. La biologie moderne essaie donc aujourd'hui d'appréhender les propriétés statistiques des évènements qui surviennent dans chacune des cellules.
Nous étudions comment le patrimoine génétique influence les lois de probabilités de ces évènements. Par des expériences mesurant des niveaux d'expression génique ou bien des caractères morphologiques de milliers de cellules individuelles, nous étudions comment les propriétés statistiques des caractères cellulaires sont modifiées par le fond génétique. Ces effets probabilistes des polymorphismes de l'ADN sont hautement pertinents pour l'interprétation de la pénétrance incomplète de certaines mutations (pourquoi une fraction des porteurs de mutations ne déclarent pas le phénotype associé).
Dans une première étude, nous avons découvert que les fonds génétiques naturels de S. cerevisiae ne génèrent pas tous le même niveau de stochasticité d'expression d'un gène rapporteur modèle. Cette stochasticité, traduite par une forte variance au sein d'une population de cellules isogéniques, est elle-même contrôlée par de multiples gènes, et les sources moléculaires de ce 'bruit' peuvent être identifiées par cartographie génétique.
|
|
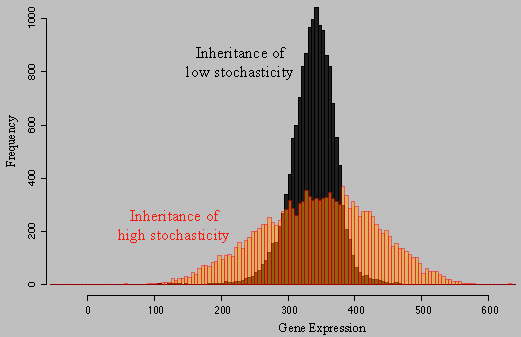
A propos de la figure: Deux fonds génétiques sauvages de S. cerevisiae ont été croisés, et les descendants de ce croisement ont été analysés afin de quantifier le niveau d'expression d'une protéine fluorescente dans 15,000 cellules individuelles. Chaque histogramme représente la distribution d'expression de MET17-GFP au sein d'une population de cellules isogéniques correspondant à un fond génétique descendant du croisement. En noir, un très faible niveau de stochasticité a été hérité, alors que le fond rouge a hérité d'un très fort niveau de stochasticité. Nous avons trouvé que l'efficacité de l'élongation de la transcription explique une partie de cette différence de variance. Pour plus de détails, merci de consulter les publications Ansel et al. 2007 et Fehrmann et al. 2013. |
Nous avons développé une nouvelle méthode pour cehrcher dans le génome des variations génétiques probabilistes. Cette méthode s'appelle PTLMAPPER. Elle est entièrement décrite dans la publication Chuffart et al. 2016 ici, et vous pouvez librement télécharger et utiliser son implémentation logicielle qui est mise à disposition sous la forme d'un package R, l'adresse est indiquée sur notre download.
Nous étudions maintenant ces effets non-déterministes des variations génétiques naturelles par des approches de Biologie des Systèmes, qui combinent des acquisitions expérimentales à haut débit avec des interprétations/prédictions basées sur des modèles mathématiques et du séquençage massif. Les tâches informatiques sont facilitées par l'accès au cluster de calcul PSMN de l'ENS de Lyon.