

2DUV and protein conformation
Les techniques de spectroscopie optique à deux dimensions basées sur des pulses lasers ultracourts ont été récemment étendues à l’UV. Les cycles aromatiques deviennent de ce fait des marqueurs pour suivre les réarrangements structuraux des protéines. Ici, nous montrons que le spectre d’un tétrapeptide modèle avec deux chaînes latérales aromatiques contient suffisamment d’informations structurales pour distinguer les configurations distante et vicinale des chaînes latérales.
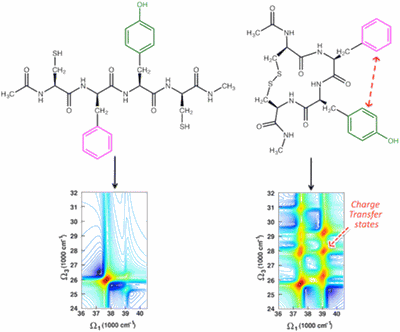
Two-dimensional (2D) optical spectroscopy techniques based on ultrashort laser pulses have been recently extended to the optical domain in the ultraviolet (UV) spectral region. UV-active aromatic side chains can thus be used as local highly specific markers for tracking dynamics and structural rearrangements of proteins. Here we demonstrate that 2D electronic spectra of a model proteic system, a tetrapeptide with two aromatic side chains, contain enough structural information to distinguish between two different configurations with distant and vicinal side chains. For accurate simulations of the 2DUV spectra in solution, we combine a quantum mechanics/molecular mechanics approach based on wave function methods, accounting for interchromophores coupling and environmental effects, with nonlinear response theory. The proposed methodology reveals effects, such as charge transfer between vicinal aromatic residues that remain concealed in conventional exciton Hamiltonian approaches. Possible experimental setups are discussed, including multicolor experiments and signal manipulation techniques for limiting undesired background contributions and enhancing 2DUV signatures of specific electronic couplings.
Artur Nenov, Ivan Rivalta, Giulio Cerullo, Shaul Mukamel, and Marco Garavelli.
Disentangling Peptide Configurations via Two-Dimensional Electronic Spectroscopy: Ab Initio Simulations Beyond the Frenkel Exciton Hamiltonian.
J. Phys. Chem. Lett., 2014, 5 (4), pp 767–771.