

Temperature-dependent NIR-CPL spectra of chiral Yb(III) complexes

L. Guy et al.
Article available at Phys. Chem. Chem. Phys.
Abstract
Chiral, enantiopure Yb(III) complexes exhibit circularly polarized luminescence (CPL) in the near infrared (NIR) wavelength region. This CPL is quantified by the dissymmetry factor (glum). The excited state 2F5/2 consists of six mJ′ states degenerated in three Stark levels, due to the crystal-field splitting (CFS), which are populated in accordance with the Boltzmann distribution. Consequently, room temperature CPL spectra are the sum of various - either positive or negative – contributions, that are practically impossible to quantify. To address this issue, an advanced setup enabling CPL measurements over a broad temperature range (300 to 4 K) has been developed. The interrelation of CFS, glum and temperature was explored using a pair of enantiopure Yb(III) complexes, highlighting the individual contribution of each crystal-field sublevel to the overall CPL spectrum, as anticipated by simulations performed in the framework of multireference wave-functions. Hence, the CPL spectra of chiral lanthanide complexes were found to be indeed strongly temperature-dependent, as is the glum dissymmetry factor, as a consequence of the variation in thermal sublevel population.
Circularly polarized luminescence activity in the near infrared spectral region of a water-soluble ytterbium(III) complex containing a conjugated chromophoric ligand

L. Guy et al.
Article available at New J. Chem.
Abstract
Two cationic enantiomeric complexes [(R,R)-[YbL]Cl and (S,S)-[YbL]Cl with L = N,N′-bis(2-pyridylmethyl)-1,2-(R,R or S,S)-cyclohexanediamine functionalized at sp3 N with picolinate antennae] have been synthesized and spectroscopically characterized in polar protic solvents, such as water and methanol. The Yb(III) luminescence at about 980 nm is efficiently sensitized upon excitation of the picolinate antenna in the UV spectral region around 330 nm. The complexes exhibit good CPL activity for the 2F5/2 → 2F7/2 magnetic dipole (MD) allowed transition (glum values of |0.02| and |0.04| at 984 nm and 1021 nm, respectively) and high solubility in aqueous solution. The YbL species is largely predominant at physiological pH (7.4), given its high thermodynamic stability (logβYbL = 20.98). As elucidated by DFT calculations, among the possible isomeric species the one characterized by the cis-O, O–N, N geometry was found to be dominant. Furthermore, one solvent molecule is bound to Ln(III) in water, giving rise to 9-fold coordination at the metal ion. The enantiomeric (R,R)-[YbL]Cl and (S,S)-[YbL]Cl complexes can be considered promising candidates as NIR-to-NIR chiroptical bioprobes also for in vivo experiments.
Annealing temperature-dependent induced supramolecular chiroptical response of copolymer thin films studied by pump-modulated transient circular dichroism spectroscopy
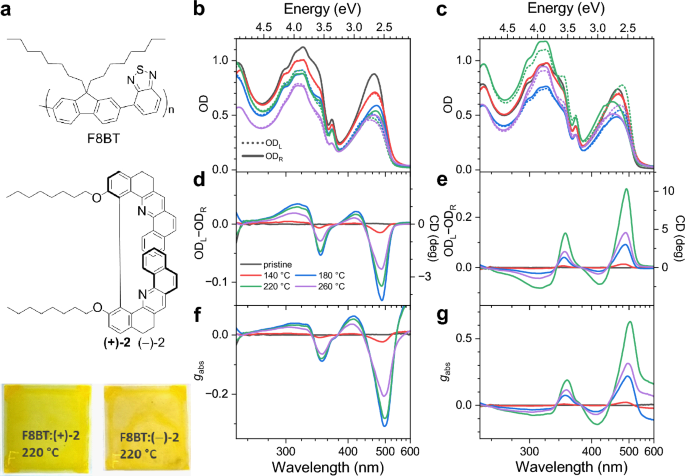
J.-C. Mulatier, D. Pitrat, L. Guy et al.
Article available at Sci Rep
Abstract
Copolymer thin films showing induced supramolecular chirality are of considerable interest for optoelectronic applications such as organic light-emitting diodes. Here, we introduce a new helicene-like chiral additive with two octyloxy substituents which displays excellent chiral induction properties in an achiral polyfluorene copolymer, leading to a circular dichroism (CD) response of up to 10,000 mdeg. This chiral inducer also displays very good thermal stability, which enables us to perform an extended study on the induced chiroptical properties of the cholesteric copolymer thin films annealed at different temperatures in the range 140–260 °C. Starting from about 180 °C, a distinct change in the morphology of the CD-active film is observed by CD microscopy, from micrometre-size granular to extended CD-active regions, where the latter ones display skewed distributions of the dissymmetry parameter gabs. Broadband Müller matrix spectroscopy finds a pronounced CD and circular birefringence (CB) response and only weak linear dichroism (LD, LD’) and linear birefringence (LB, LB’). Ultrafast transient CD spectroscopy with randomly polarised excitation reveals a clean mirror-image-type transient response, which shows a second-order decay of the S1 population due to singlet–singlet annihilation processes.
Chiroptical Properties of a Cryptophane Soluble at Neutral pH

T. Brotinet al.
Article available at Eur. J. Org. Chem.
Abstract
This article reports the synthesis of the two enantiomers of a water-soluble cryptophane 3 enabling cesium and thallium complexation at neutral or basic pH. The two enantiomers of 3 were obtained in a two steps synthesis from cryptophane 4, a molecule decorated with three phenol groups and three other phenol groups protected by benzyl units. The two enantiomers of 4 were isolated from liquid chromatography using chiral stationary phase. The absolute configuration of the two enantiomers of 3 was determined by vibrational circular dichroism combined with theoretical calculations. Contrary to cryptophane 1 that present large spectroscopic changes upon cesium and thallium complexation, the electronic circular dichroism spectra of compound 3 exhibit moderate spectral changes suggesting that this compound cannot change the conformation of its linkers as easily as cryptophane 1.
Understanding the surrounding effects on Raman optical activity signatures of a chiral cage system: Cryptophane-PP-111

D. Pitrat, T. Brotinet al.
Article available at Spectrochimica Acta Part A
Abstract
Cryptophane molecules are cage-like structures consisting in two hemispheres, each made of three benzene rings. These hemispheres are bound together with three single bondO(CH2) Osingle bondlinkers of various lengths giving rise to a plethora of cryptophane derivatives. Moreover, they are able to encapsulate neutral guests: CH2Cl2, CHCl3, …; and charged species: Cs+, Tl+, …. Finally, they exhibit chiroptical properties thanks to the anti arrangement of the linkers between the hemispheres. This work focuses on the Raman optical activity (ROA) signatures of Cryptophane-111 ( for each linker). More specifically, we aim at simulating accurately its ROA spectra with and without a xenon atom inside its cavity. Experimental data (Buffeteau et al., 2017) have already demonstrated the effect of the encapsulation in the low-wavenumbers region. To generate the initial structures, we rely on the novel Conformer–Rotamer Ensemble Sampling Tool (CREST) program, developed by S. Grimme and co-workers. This is required due to the flexibility provided by the linkers. The CREST algorithm seems promising and has already been used to sample the potential energy surface (PES) of target systems before the simulation of their vibrational spectroscopies (Eikås et al., 2022). We observe large similarities between the two sets of conformers (one with and one without Xe encapsulated), demonstrating the robustness of the CREST algorithm. For corresponding structures, the presence of xenon pushed the two hemispheres slightly further apart. After optimization at the DFT level, only one unique conformer has a Boltzmann population ratio greater than 1%, pointing out the relative rigidity of the cage. Based on this unique conformer, our simulations are in good agreement with the experimental data. Regarding xenon encapsulation, the (experimental and theoretical) ROA signatures at low wavenumbers are impacted: slight shifts in wavenumbers are observed as well as a decrease in relative ROA intensity for bands around 150 cm . The wavenumber shifts were very well reproduced by our simulations, but the experimental decrease in the ROA intensity was unfortunately not reproduced.
A Water-Soluble Cryptophane Decorated with Aromatic Amine Groups Shows High Affinity for Cesium and Thallium(I)

R. Benchouaia, M. Doll, T. Brotin,N. De Rycke et al.
Article available at J. Org. Chem.
Abstract
An anti-cryptophane decorated with three aromatic amine and three phenol groups shows a high affinity for the cesium and thallium cations in LiOH/H2O (0.1 M). The formation of the complexes was studied by 133Cs NMR and by 205Tl NMR spectroscopy at different temperatures. Characteristic signals for caged cesium and thallium were observed at a high field with respect to the signals of the free cations present in the bulk. Isothermal titration calorimetric experiments performed in LiOH/H2O (0.1 M) and NaOH/KCl buffer (pH = 13) allowed us to determine the parameter of complexation and to ascertain the high affinity of this cryptophane for cesium and thallium. A comparison with other cryptophanes that bind these two cations shows that the introduction of nitrogen atoms into the cryptophane backbone has an effect on the binding properties. The affinity for cesium and thallium(I) ions is in the following order of substitution: OH > NH2 > OCH2COOH. This study paves the way to the design of new efficient host molecules for the extraction of these two cations in aqueous solution.
Pizza Oven Processing of Organohalide Perovskites (POPOP): A Simple, Versatile and Efficient Vapor Deposition Method

C. Bucher et al.
Article available at Adv. Energy Mater.
Abstract
Hybrid vapor deposition is one of the most appealing processes for perovskite photovoltaics fabrication, thanks to its versatile nature. By using sequentially different vapor deposition processes tailored to the inorganic and organic perovskite precursors' peculiarities, this type of process gives access to the full potential of vapor deposition. While vapor deposition of metal halides is well understood and mastered, vapor deposition of organohalide species is much more delicate (degradation of vapors, high vapor pressure, setup-specific constraints). Here, a novel close space sublimation system is reported and in-depth insights on the conversion into perovskite of a metal halide template are provided. In this evolution of the process, the substrate coated with metal halide template and the organohalide source are loaded together in a dedicated holder, then transferred into a vacuum chamber on a heating element already at temperature setpoint. The system enables a simple, fast, low-cost, and easy-to-reproduce organohalide vapor deposition process. The formation of the perovskite in situ and identification different conversion regimes are studied. Furthermore, the influence of the chemical environment and chamber design on the process are discussed. Compositional tuning and additive engineering in the process are processed and fabricate proof of concept photovoltaic devices reaching high fill factors of 80% and 17% power conversion efficiency for a bandgap of 1.63 eV.
Electron-Triggered Imine Coupling: Synthesis and Characterization of Three Redox States (0,–1,–2) of a Ni(N2S2) Complex

C. Bucher et al.
Article available at Chem. Eur. J. .
Abstract
Metal imine-thiolate complexes, M(NS)2 are known to undergo imine C−C bond formation to give M(N2S2) complexes (M=Co, Ni) containing a redox-active ligand. Although these transfor-mations are not typically quantitative, we demonstrate here that the one-electron reduction of a related Ni bis(imine-thiolate) complex affords the corresponding paramagnetic [Ni(N2S2)]- anion (2⋅−) exclusively; subsequent oxidation with [Cp2Fe]BF4 then affords a high yield of neutral 2 (Cp=η5-cyclopentadienyl). Moreover, electrochemical studies indicate that a second one-electron reduction affords the diamagnetic dianion. Both anionic products were isolated and characterized by SC-XRD and their electronic structures were investigated by UV-vis spectro-electrochemistry, EPR and NMR spectroscopy, and DFT studies. These studies show that reduction proceeds primarily on the ligand, with (N2S2)4− containing both thiolate and ring-delocalized anions
Unprecedented light induced aggregation of cationic 1,4,5,8-naphthalenediimide amphiphiles

D. Frath, C. Bucher et al.
Article available at J. Mater. Chem. C..
Abstract
Naphthalenediimide amphiphiles (NDI-as) with quaternary ammonium groups (DC4, DaP, and DaO) display unprecedented UV light-induced aggregation in solutions of water, acetonitrile and THF. Light-induced aggregation brings significant spectral modification by which unusual absorption band distribution is assigned to H-type aggregates. Such aggregation can be reversibly broken by heating and reassembled by photoirradiation. The irradiation/heating cycles that provoke assembly/disassembly process can be confirmed by 1H-NMR spectroscopy. The possibility of a chemically promoted aggregation by or radical anion either anion–π interaction may be ruled out by UV-Vis, EPR and electrochemistry measurements. These photoinduced aggregates can sensitize dissolved O2 to produce singlet oxygen, a surprising observation when it comes to sensitization efficiency for dyes as aggregation usually decreases sensitization yield. Laser-induced luminescence measurements corroborate the existence of monomeric and aggregate forms, as well as confirm triplet states as phosphorescence was detected, being particularly intense in concentrated THF solutions.
Phosphonate-substituted porphyrins as efficient, cost-effective and reusable photocatalysts

C. Bucher et al.
Article available at Dalton Trans..
Abstract
Recent advances in visible light photocatalysis represent a significant stride towards sustainable catalytic chemistry. However, its successful implementation in fine chemical production remains challenging and requires careful optimization of available photocatalysts. Our work aims to structurally modify bioinspired porphyrin catalysts, addressing issues related to their laborious synthesis and low solubility, with the goal of increasing their efficiency and developing reusable catalytic systems. We have demonstrated the catalytic potential of readily available meso-tetrakis[4-(diethoxyphosphoryl)phenyl]porphyrins (M(TPPP)). Novel metal (Pd(II), Co(II) and In(III)) complexes with this ligand were prepared in good yields. These chromophores were characterized in solution using spectroscopic (NMR, UV–vis, fluorescence) and electrochemical methods. The introduction of phosphonate groups on the phenyl substituents of meso-tetraphenylporphyrins (M(TPP)) improves solubility in polar organic solvents without significantly altering the photophysical properties and photostability of complexes. This structural modification also leads to easier reductions and harder oxidations of the macrocycle for all investigated complexes compared to the corresponding TPP derivatives. The free base porphyrin, zinc(II), palladium(II), and indium(III) complexes were studied as photocatalysts for oxidation of sulfides to sulfoxides using molecular oxygen as a terminal oxidant. Both dialkyl and alkyl aryl sulfides were quantitatively transformed into sulfoxides under blue LED irradiation in the acetonitrile–water mixture (10 : 1 v/v) with a low loading (0.005–0.05 mol%) of porphyrin photocatalysts, where H2(TPPP) and Pd(TPPP) were found to be the most efficient. The reaction mechanism was studied using photoluminescence and EPR spectroscopies. Then, to access reusable catalysts, water-soluble derivatives bearing phosphonic acid groups, H2(TPPP-A) and Pd(TPPP-A), were prepared in high yields. These compounds were characterized using spectroscopic methods. Single-crystal X-ray diffraction analysis of Pd(TPPP-A) reveals that the complex forms a 3D hydrogen-bonded organic framework (HOF) in the solid state. Both H2(TPPP-A) and Pd(TPPP-A) were found to catalyze the photooxidation of sulfides by molecular oxygen in the acetonitrile–water mixture (1 : 1 v/v), while only Pd(TPPP-A) resulted in selective production of sulfoxides. The complex Pd(TPPP-A) was easily recovered through extraction in the aqueous phase and successfully reused in five consecutive cycles of the sulfoxidation reaction..
Chemically Mediated Artificial Electron Transport Chain

C. Bucher et al.
Article available at ACS Cent. Sci.
Abstract
Electron transport chains (ETCs) are ubiquitous in nearly all living systems. Replicating the complexity and control inherent in these multicomponent systems using ensembles of small molecules opens up promising avenues for molecular therapeutics, catalyst design, and the development of innovative energy conversion and storage systems. Here, we present a noncovalent, multistep artificial electron transport chains comprising cyclo[8]pyrrole (1), a meso-aryl hexaphyrin(1.0.1.0.1.0) (naphthorosarin 2), and the small molecules I2 and trifluoroacetic acid (TFA). Specifically, we show that 1) electron transfer occurs from 1 to give I3– upon the addition of I2, 2) proton-coupled electron transfer (PCET) from 1 to give H32•2+ and H32+ upon the addition of TFA to a dichloromethane mixture of 1 and 2, and 3) that further, stepwise treatment of 1 and 2 with I2 and TFA promotes electron transport from 1 to give first I3– and then H32•2+ and H32+. The present findings are substantiated through UV-vis-NIR, 1H NMR, electron paramagnetic resonance (EPR) spectroscopic analyses, cyclic voltammetry studies, and DFT calculations. Single-crystal structure analyses were used to characterize compounds in varying redox states.