

Ammonia Synthesis at Room Temperature and Atmospheric Pressure from N2: A Boron-Radical Approach

C. Bucher et al.
Article available at ACIE
Abstract
Ammonia, NH3, is an essential molecule, being part of fertilizers. It is currently synthesized via the Haber–Bosch process, from the very stable dinitrogen molecule, N2 and dihydrogen, H2. This process requires high temperatures and pressures, thereby generating ca 1.6 % of the global CO2 emissions. Alternative strategies are needed to realize the functionalization of N2 to NH3under mild conditions. Here, we show that boron-centered radicals provide a means of activating N2 at room temperature and atmospheric pressure whilst allowing a radical process to occur, leading to the production of borylamines. Subsequent hydrolysis released NH4+, the acidic form of NH3. EPR spectroscopy supported the intermediacy of radicals in the process, corroborated by DFT calculations, which rationalized the mechanism of the N2functionalization by R2B radicals.
Synthesis and Chiroptical Properties of a Chiral Isotopologue of syn-Cryptophane-B
T. Brotin et al.
Article available at J. Org. Chem
Abstract
We report the synthesis and absolute configuration (AC) of a chiral isotopologue of syn-cryptophane-B. Low chiral signatures were measured by polarimetry and electronic circular dichroism, whereas most significant chiroptical effects were observed by vibrational circular dichroism (VCD) and Raman optical activity (ROA). The comparison of experimental VCD and ROA spectra with those predicted by DFT calculations allows the determination of the AC of the two enantiomers as (−)589-MP-syn-2 and (+)589-PM-syn-2.
Acid/Base-Triggered Photophysical and Chiroptical Switching in a Series of Helicenoid Compounds
L. Guy et al.
Article available at Molecules.
Abstract
Internal Dynamics and Modular Peripheral Binding in Stimuli-Responsive 3 : 2 Host:Guest Complexes

F. Chevalier, C. Bucher et al.
Article available at ACIE.
Abstract
Now that the chemistry of 1 : 1 host:guest complexes is well-established, it is surprising to note that higher stoichiometry (oligomeric) complexes, especially those with excess host, remain largely unexplored. Yet, proteins tend to oligomerize, affording new functions for cell machinery. Here, we show that cucurbit[n]uril (CB[n]) macrocycles combined with symmetric, linear di-viologens form unusual 3 : 2 host:guest complexes exhibiting remarkable dynamic properties, host self-sorting, and external ring-translocation. These results highlight the structural tunability of cucurbit[8]uril (CB[8]) based 3 : 2 host:guest complexes in water and their responsiveness toward several stimuli (chemicals, pH, redox).
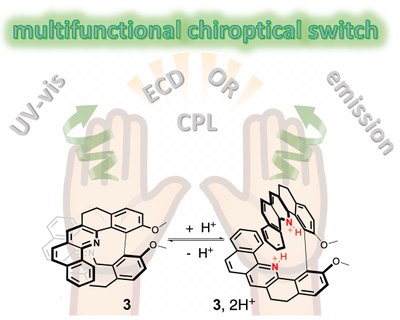
A series of molecules that possess two quinolines, benzoquinolines, or phenanthrolines connected in a chiral fashion by a biaryl junction along with their water-soluble derivatives was developed and characterized. The influence of the structure on the basicity of the nitrogen atoms in two heterocycles was examined and the photophysical and chiroptical switching activity of the compounds upon protonation was studied both experimentally and computationally. The results demonstrated that changes in the electronic structure of the protonated vs. neutral species, promoting a bathochromic shift of dominant electronic transitions and alternation of their character from 𝜋-to-𝜋* to charge-transfer-type, when additionally accompanied by the high structural flexibility of a system, leading to changes in conformational preferences upon proton binding, produce particularly pronounced modifications of the spectral properties in acidic medium. The latter combined with reversibility of the read-out make some of the molecules in this series very promising multifunctional pH probes.
Proton-coupled electron transfer in a pivaloyl-substituted dihydro-tetraazapentacene

C. Bucher et al.
Article available at Electrochemica acta
Abstract
Within the heteroacene family, azapentacenes have received much attention in organic electronic devices due to their unique physico-chemical properties. This is particularly true for the 5,14-dihydro-5,7,12,14-tetraazapentacene despite its poor solubility in organic solvents which logically hindered the study of its electrochemical behavior. Consequently, we disclose herein the synthesis and characterization of the soluble 6,13-dipivaloyl-5,14-dihydro-5,7,12,14-tetraazapentacene as well as its aromatic counterpart, the 6,13-dipivaloyl-5,7,12,14-tetraazapentacene. These compounds were used as models to fully characterize all the intermediates generated during their electrochemical processes (oxidation and reduction) with a combined approach using complementary chemical, (spectro)-electrochemical, theoretical and spectroscopic techniques. Our results demonstrate the 6,13-dipivaloyl-5,14-dihydro-5,7,12,14-tetraazapentacene ambipolar redox behavior at both cathodic and anodic regime leading to drastic color changes through proton-coupled electron transfer occuring in a totally reversible fashion.
Supramolecular polymers: Recent advances based on the types of underlying interactions

C. Bucher et al.
Article available at Progress in Polymer Science
Abstract
Supramolecular polymers are, in broad brushstrokes, self-assembled structures built up from small building blocks via the use of noncovalent interactions. In favorable cases, supramolecular polymers embody the best features of covalent polymers while displaying unique reversibility, responsiveness, adaptiveness, and stability. This has made them of interest across a wide variety of fields, from molecular devices to sensors, drug delivery, cell recognition, and environmentally friendly materials systems. This review is concerned with the determinants that underlie supramolecular polymer construction, specifically the driving forces that have been exploited to create them. To date, nearly the full range of known noncovalent interactions (e.g., hydrogen-bonding, electrostatic interactions, charge transfer effects, and metal coordination, among others) has been exploited to create supramolecular polymers. Typically, one or more types of interactions is used to link appropriately designed monomers. The choice of noncovalent interaction can have a significant influence on the structure and function of the resulting supramolecular polymers. Understanding the connections between the forces responsible for the assembly of supramolecular polymers and their properties provides the foundation for further advances in this fast-moving field. Given the above, this review will discuss recent progress in the rapidly advancing field of supramolecular polymers organized by the types of underlying interactions. An overview of future challenges and opportunities for supramolecular polymers, including their formation, characterization, and applications, is also provided.
Subtle Stereochemical Effects Influence Binding and Purification Abilities of an FeII4L4 Cage

T. Brotin et al.
Article available at JACS
Abstract
A tetrahedral FeII4L4 cage assembled from the coordination of triangular chiral, face-capping ligands to iron(II). This cage exists as two diastereomers in solution, which differ in the stereochemistry of their metal vertices, but share the same point chirality of the ligand. The equilibrium between these cage diastereomers was subtly perturbed by guest binding. This perturbation from equilibrium correlated with the size and shape fit of the guest within the host; insight as to the interplay between stereochemistry and fit was provided by atomistic well-tempered metadynamics simulations. The understanding thus gained as to the stereochemical impact on guest binding enabled the design of a straightforward process for the resolution of the enantiomers of a racemic guest.
Allosterically Regulated Guest Binding Determines Framework Symmetry for an FeII4L4 Cage

T. Brotin et al.
Article available at ACIE
Abstract
Self-assembly of a flexible tritopic aniline and 3-substituted 2-formylpyridine subcomponents around iron(II) templates gave rise to a low-spin FeII4L4 capsule, whereas a high-spin FeII3L2 sandwich species formed when a sterically hindered 6-methyl-2-formylpyridine was used. The FeII4L4 cage adopted a new structure type with S4symmetry, having two mer-Δ and two mer-Ʌ metal vertices, as confirmed by NMR and X-ray crystallographic analysis. The flexibility of the face-capping ligand endows the resulting FeII4L4 framework with conformational plasticity, enabling it to adapt structurally from S4 to T or C3 symmetry upon guest binding. The cage also displayed negative allosteric cooperativity in simultaneously binding different guests within its cavity and at the apertures between its faces.
Al(III) and Ga(III) Bisphenolate Azadipyrromethene-Based “N2O2” Complexes as Efficient NIR-Fluorophores

C. Bucher et al.
Article available at Inorg. Chem.
Abstract
Aza-boron-dipyrromethenes (Aza-BODIPYs) are an increasingly studied class of fluorophores. They can be seen as an azadipyrromethene (“aza-DIPY”) ligand rigidified by a metalloid, a boron atom. Based on this idea, a series of complexes of group 13 metals (aluminum and gallium) have been synthesized and characterized. The impact of the metal and of the nature of the substituents of aza-DIPY core were investigated. The photophysical and electrochemical properties were determined, and an X-ray structure of an azaGaDIPY was obtained. These data reveal that azaGaDIPY and azaAlDIPY exhibit significant red-shifted fluorescence compared to their analogue aza-BODIPY. Their emission can go up to 800 nm for the maximum emission length and up to NIR-II for the emission tail. This, associated with their electrochemical stability (no metal release whether oxidized or reduced) makes them a promising class of fluorophores for optical medical imaging. Moreover, X-ray structure and molecular modeling studies have shown that this redshift seems to be more due to the geometry around the boron/metal than to the nature of the metal.
Tuning the Photophysical Properties of Aza-BODIPYs in the Near-Infrared Region by Introducing Electron-Donating Thiophene Substituents

C. Bucher et al.
Article available at Chem. Eur. J.
Abstract
This study presents the synthesis, the spectroscopic and electrochemical properties of new bis- and tetra-substituted azaboron-dipyrromethene (aza-BODIPY) dyes substituted by different electron donating groups connected to the aza-BODIPY core through a thiophene unit. In line with theoretical calculations, experimental measurements point out the positive impact of the thiophene group that behave as a secondary donor group leading to an enhancement of the intramolecular charge transfer process in comparison to previously reported aza-BODIPY dyes. This heterocycle has also been found to tune the oxidative potential and to stabilize the electro-generated species.
Electron-Triggered Imine Coupling: Synthesis and Characterization of Three Redox States (0,–1,–2) of a Ni(N2S2) Complex

C. Bucher et al.
Article available at Chem. Eur. J.
Abstract
Metal imine-thiolate complexes, M(NS)2 are known to undergo imine C−C bond formation to give M(N2S2) complexes (M=Co, Ni) containing a redox-active ligand. Although these transfor-mations are not typically quantitative, we demonstrate here that the one-electron reduction of a related Ni bis(imine-thiolate) complex affords the corresponding paramagnetic [Ni(N2S2)]- anion (2⋅−) exclusively; subsequent oxidation with [Cp2Fe]BF4then affords a high yield of neutral 2 (Cp=η5-cyclopentadienyl). Moreover, electrochemical studies indicate that a second one-electron reduction affords the diamagnetic dianion. Both anionic products were isolated and characterized by SC-XRD and their electronic structures were investigated by UV-vis spectro-electrochemistry, EPR and NMR spectroscopy, and DFT studies. These studies show that reduction proceeds primarily on the ligand, with (N2S2)4− containing both thiolate and ring-delocalized anions.
Chiral and conductive viologen-based supramolecular gels exhibiting tunable charge-transfer properties
V. Andrieux, L. Guy, F. Chevallier, D. Frath, C. Bucher et al.
Article available at J. Mater. Chem. C.
Abstract
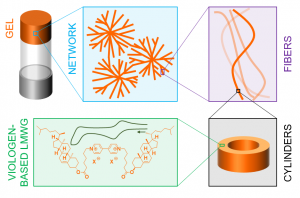
Redox-active conductive supramolecular gels involving highly ordered chiral assemblies of small organic molecules are very promising soft materials for many applications ranging from catalysis to electronics. However, combining all these properties in the same material has so far remained a difficult task. We now report the synthesis and detailed structural, rheological, and electrical characterizations of supramolecular gels obtained by self-assembly of a dicationic low molecular weight gelator incorporating a redox-active 4,4′-bipyridinium unit. These molecules have been shown to self-assemble in pentanol to form chiral hollow core–shell cylinders, eventually yielding dendritic clusters inducing gelation. We also showed that the optical, rheological, and electrical properties of the gels can be tuned by adding ionic additives. Careful control of the formation of charge-transfer complexes between viologens and iodides has led to the formation of robust, transparent, conductive, and chiral gel. The gelation process, the properties of the gel, and the structure of the assemblies have been thoroughly investigated by UV-Vis and ECD spectroscopy, rheometry, bright-field microscopy, SAXS, AFM, electrochemical and impedance measurements.
Redox-responsive 1D-assembly built from cucurbit[8]uril and a water-soluble metalloporphyrin-based tecton
S. Chowdhury, P. Hennequin, S. Denis-Quanquin, D. Frath, F. Chevallier, C. Bucher et al.
Article available at J. Porphyr. Phthalocyanines.
Abstract
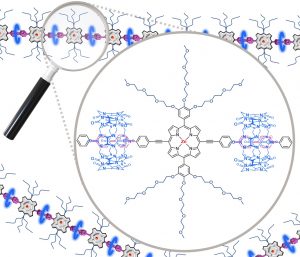
A linear porphyrin-based tecton bearing two 4,4′bipyridinium units (viologens) and two monomethyl-ether triethylene glycol-substituted phenyl substituents at the meso positions was synthesized and characterized. The latter was involved in the redox-triggered formation of linear supramolecular assemblies with cucurbit[8]uril (CB[8]) cavitands in aqueous media. The CB[8]-promoted intermolecular π-dimerization of the viologen cation radicals introduced at the meso positions of the porphyrin platform has been brought to light through the diagnostic signatures of the 1:2 host-guest ternary caviplexes formed between viologen and CB[8] and by spectroscopic data collected after electrochemical reduction of the viologen-based tectons.
Theoretical and experimental analysis of CPL spectrophotometers for artifact-free measurements using a single CCD camera
L. Guy et al.
Article available at Nat. Commun.
Abstract
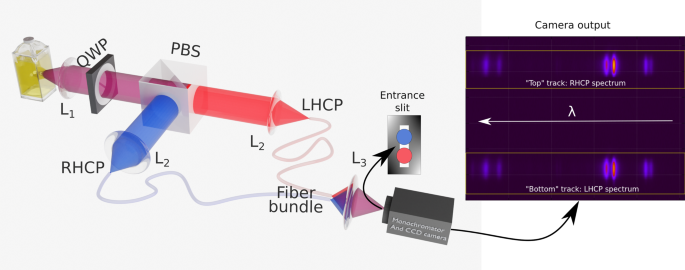
Circularly polarized luminescence (CPL) is a fast growing research field as a complementary chiroptical spectroscopy alternative to the conventional circular dichroism or in the quest of devices producing circularly polarized light for different applications. Because chiroptical signals are generally lower than 0.1%, conventional chiral spectroscopies rely on polarization time modulation requiring step-by-step wavelength scanning and a long acquisition time. High throughput controls motivated the development of CPL spectrophotometers using cameras as detectors and space polarization splitting. However, CPL measurements imposes careful precautions to minimize the numerous artifacts arising from experimental imperfections. Some previous work used complex calibration procedure to this end. Here we present a rigorous Mueller analysis of an instrument based on polarizations space splitting. We show that by using one camera and combining spatial and temporal separation through two switchable circular polarization encoding arms we can record accurate CPL spectra without the need of any calibration. The measurements robustness and their fast acquisition times are exemplified on different chiral emitters.
Circularly polarized activity from two photon excitable europium and samarium chiral bioprobes

L. Guy et al.
Article available at J. Mater. Chem. C.
Abstract
In this contribution, two couples of cationic enantiomeric complexes [(R,R)-[LnL]Cl and (S,S)-[LnL]Cl, with Ln = Sm and Eu and L = N,N′-bis(2-pyridylmethyl)-1,2-(R,R or S,S)-cyclohexanediamine functionalized at sp3 N with the picolinate antennae2] have been synthesized and spectroscopically characterized in polar protic solvents, such as water and methanol. The good sensitization of Sm(III) and Eu(III) luminescence by the picolinate antenna is documented, upon both a one photon absorption process (at about 330 nm) and a two photon absorption process (at 720 nm). The complexes exhibit good CPL activity, in particular for the magnetic dipole (MD) allowed transitions, which are the 4G5/2 → 6H5/2 (564 nm) of Sm(III) and the 5D0 → 7F1 (593 nm) of Eu(III). Both complexes are highly stable in aqueous solution (log K = 20.13 for the EuL species chosen as the representative) and only one main species is present at physiological pH (7.4). Among the three possible isomeric complexes, DFT calculations on the Y(III) counterpart reveal the highest stability of the C1-symmetric cis–O, O–N, N isomer, in agreement with the presence of only one Eu(III) emitting species in solution. Furthermore, one solvent molecule is bound to the metal ions, giving rise to 9-fold coordinated complexes. Preliminary biphotonic imaging experiments on the (S,S)-[EuL]Cl complex reveal that it can be easily internalized in two different cell lines (namely 293T cancer cells and THP-1 macrophages) with perinuclear diffuse localisation in the cytosol. Both Sm(III) and Eu(III) complexes can be considered promising candidates as NIR-to-RED in cellulo chiroptical bioprobes and a possible extension to the in vivo experiment will be further investigated.
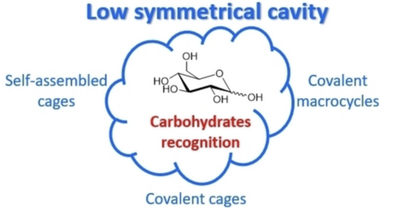
Low-Symmetry Macrocycles and Cages for Carbohydrate Recognition
J.-P. Dutasta et al.
Article available at ChemPlusChem
Abstract
The recognition of carbohydrate plays a key role in numerous biological processes. Thus, artificial receptors have been synthesized to mimic these biological systems. To date, most of the receptors reported for carbohydrate complexation present highly symmetrical cavities, probably because their syntheses require less synthetic efforts and are easier to achieve and control. However, carbohydrates display complex, asymmetrical structures suggesting that hosts with low symmetry might be more adapted to recognize these guests. Here, we described the strategies that have been used to complex carbohydrates with macrocycles and cages presenting low symmetry and the potential of this approach. Self-assembled cages are first described, then covalent macrocycles and cages are presented and for each example the binding properties of low-symmetry systems are compared to those of their higher-symmetry counterparts.
Unprecedented Relaxivity Gap in pH-Responsive FeIII-Based MRI Probes
J. Salaam, T. Fogeron, C. Bucher, L. Khrouz, J. Hasserodt et al.
Article available at Angew. Chem.
Abstract
Two mononuclear ferric complexes are reported that respond to a pH change with a 27- and 71-fold jump, respectively, in their capacity to accelerate the longitudinal relaxation rate of water-hydrogen nuclei, and this starting from a negligible base value of only 0.06. This unprecedented performance bodes well for tackling the sensitivity issues hampering the development of Molecular MRI. The two chelates also excel in the fully reversible and fatigue-less nature of this phenomenon. The structural reasons for this performance reside in the macrocyclic nature of the hexa-dentate ligand, as well as the presence of a single pendant arm displaying a five-membered lactam or carbamate which show (perturbed) pKavalues of 3.5 in the context of this N6 N5O1 coordination motif.
N5O1 coordination motif.
Fluorescence Detection of the Persistent Organic Pollutant Chlordecone in Water at Environmental Concentrations

J.-P. Dutasta et al.
Article available at Chem. Eur. J.
Abstract
Chlordecone (CLD), a Persistent Organic Pollutant, is still present in water and food chain of the French West Indies (FWI), leading to dramatical public health problems. One of the major issues is the lack of an easy, non-expensive, sensitive and robust method for the detection of chlordecone to ensure chlordecone-free water and foods for the residents of the FWI. This study reports on the development of a fluorescent molecular cage that allows a simple and convenient detection of chlordecone in water at environmental concentration. The specific structural features of chlordecone prompted the choice of hemicryptophanes as receptor. First, the size, shape of the cavity, as well as the recognition units, were optimized to identify the most efficient non fluorescent host for CLD complexation. Then, this selected compound was equipped with fluorophores at different positions in order to find the most efficient system for CLD detection by fluorescence. Among the two most promising fluorescent cages, the newly synthesized hemicryptophane with biphenyl moieties allowed the development of a fast, easy, reproducible and cheap procedure to detect CLD in water. Based on its sensitivity and scalability, with modulation of hemicryptophane, concentration, CLD concentrations were estimated over five orders of magnitude (10−2–103 μg/L) including the environmental levels of contamination and the permissible limit for drinking water in the FWI.
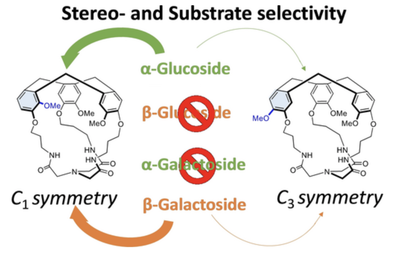
Probing the Importance of Host Symmetry on Carbohydrate Recognition
J.-P. Dutasta et al.
Article available at Chem. Eur. J.
Abstract
The design of molecular cages with low symmetry could allow for more specific tuning of their properties and better mimic the unsymmetrical and complex environment of protein pockets. However, the added value of lowering symmetry of molecular receptors has been rarely demonstrated. Herein, C3- and C1-symmetrical cages, presenting the same recognition sites, have been synthesized and investigated as hosts for carbohydrate recognition. Structurally related derivatives of glucose, galactose and mannose were found to have greater affinity to the receptor with the lowest symmetry than to their C3-symmetrical analogue. According to the host cavity modelling, the C1symmetry receptor exhibits a wider opening than its C3-symmetrical counterpart, providing easier access and thus promoting guest proximity to binding sites. Moreover, our results show the high stereo- and substrate selectivity of the C1 symmetry cage with respect to its C3 counterpart in the recognition of sugars.