





Publication du labo de chimie
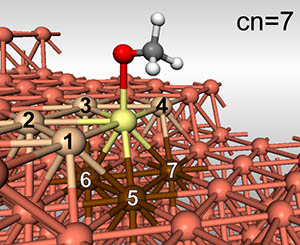
Surface d’un catalyseur métallique. On distingue : un réactif contenant un oxygène (en rouge), le site métallique considéré (en jaune) et son nombre de voisins obtenu par simple comptage. © David Loffreda
Prévoir si un catalyseur métallique sera a priori efficace nécessite des efforts de calcul considérables. De nouvelles lois appelées loi d’échelle, simplifiant grandement cette tâche, sont maintenant disponibles. Des chercheurs de l’Institut de chimie de Lyon (CNRS / Université de Lyon/ Université Claude Bernard Lyon1 et ENS de Lyon) et de l’Université de Leiden ont franchi une nouvelle étape en rendant ces lois d’échelles sensibles à la géométrie du site actif du catalyseur, permettant ainsi de prédire de manière encore plus fiable l’efficacité d’un catalyseur pour une réaction donnée. Ces travaux font l’objet d’un article dans la revue Nature Chemistry.
Bien que la catalyse soit aussi ancienne que la chimie et que quelques principes généraux aient été énoncés dès le début du XXe Siècle, la compréhension de son fonctionnement fait toujours l'objet d'activités de recherche intenses. Les catalyseurs métalliques sont susceptibles de jouer un rôle clé dans la protection de l’environnement, dans la chimie verte et dans les processus de conversion ou de stockage de l’énergie ; on comprend donc l’importance de pouvoir en inventer de nouveaux, toujours plus actifs et sélectifs dans la formation des molécules souhaitées. Le développement de catalyseurs plus performants nécessite à la fois des études de chimie expérimentale et des approches de chimie théorique, amenant une meilleure compréhension des phénomènes en œuvre et permettant parfois de prédire de nouvelles formules catalytiques. Sans étude théorique, le développement de catalyseurs pourrait être réduit à une recherche aveugle.
–
Les catalyseurs solides sont actifs par leur surface. Cependant, comme l’illustre la figure ci-dessus, les atomes de surface sont bien moins nombreux que ceux du cœur du catalyseur. L’étude expérimentale de la surface est donc délicate car celle-ci ne représente qu’une partie infime de l’échantillon. L’étude théorique se pose ainsi comme un recours à la compréhension de l’activité catalytique, d’autant que les outils permettant cette exploration ont acquis un droit légitime de cité. La question qui se pose est de trouver des critères qui permettent de prévoir si un catalyseur sera efficace ou pas, a priori.
Lire l'article intégral sur le site de l'Institut de chimie du CNRS
(...) Les chercheurs lyonnais ont franchi une nouvelle étape en rendant ces lois d’échelles sensibles à la géométrie du site actif du catalyseur, les généralisant ainsi. Ils ont montré que la géométrie du site de surface peut être simplement prise en compte en considérant le nombre de voisins de l'atome métallique où la réaction a lieu (cn = nombre de coordination, dans la figure). Ces lois d'échelles généralisées permettent donc de prédire comment choisir à la fois la composition chimique et la structure de surface d’un catalyseur afin de le rendre optimal pour une réaction donnée.
Ce travail a été développé et financé dans le cadre du contrat Européen PUMA MIND (Grant n°303419; Call FCH-JU-2011-1; Code SP1-JTI-FCH.2011.1.3).
Référence : Federico Calle-Vallejo, David Loffreda, Marc T. M. Koper and Philippe Sautet, Introducing structural sensitivity into scaling relations between adsorption energies by means of coordination numbers, Nature Chemistry 6 avril 2015, DOI : 10.1038/NCHEM.2226
Prévoir si un catalyseur métallique sera a priori efficace nécessite des efforts de calcul considérables. De nouvelles lois appelées loi d’échelle, simplifiant grandement cette tâche, sont maintenant disponibles. Des chercheurs de l’Institut de chimie de Lyon (CNRS / Université de Lyon/ Université Claude Bernard Lyon1 et ENS de Lyon) et de l’Université de Leiden ont franchi une nouvelle étape en rendant ces lois d’échelles sensibles à la géométrie du site actif du catalyseur, permettant ainsi de prédire de manière encore plus fiable l’efficacité d’un catalyseur pour une réaction donnée. Ces travaux font l’objet d’un article dans la revue Nature Chemistry.
Bien que la catalyse soit aussi ancienne que la chimie et que quelques principes généraux aient été énoncés dès le début du XXe Siècle, la compréhension de son fonctionnement fait toujours l'objet d'activités de recherche intenses. Les catalyseurs métalliques sont susceptibles de jouer un rôle clé dans la protection de l’environnement, dans la chimie verte et dans les processus de conversion ou de stockage de l’énergie ; on comprend donc l’importance de pouvoir en inventer de nouveaux, toujours plus actifs et sélectifs dans la formation des molécules souhaitées. Le développement de catalyseurs plus performants nécessite à la fois des études de chimie expérimentale et des approches de chimie théorique, amenant une meilleure compréhension des phénomènes en œuvre et permettant parfois de prédire de nouvelles formules catalytiques. Sans étude théorique, le développement de catalyseurs pourrait être réduit à une recherche aveugle.
–
Les catalyseurs solides sont actifs par leur surface. Cependant, comme l’illustre la figure ci-dessus, les atomes de surface sont bien moins nombreux que ceux du cœur du catalyseur. L’étude expérimentale de la surface est donc délicate car celle-ci ne représente qu’une partie infime de l’échantillon. L’étude théorique se pose ainsi comme un recours à la compréhension de l’activité catalytique, d’autant que les outils permettant cette exploration ont acquis un droit légitime de cité. La question qui se pose est de trouver des critères qui permettent de prévoir si un catalyseur sera efficace ou pas, a priori.
Lire l'article intégral sur le site de l'Institut de chimie du CNRS
(...) Les chercheurs lyonnais ont franchi une nouvelle étape en rendant ces lois d’échelles sensibles à la géométrie du site actif du catalyseur, les généralisant ainsi. Ils ont montré que la géométrie du site de surface peut être simplement prise en compte en considérant le nombre de voisins de l'atome métallique où la réaction a lieu (cn = nombre de coordination, dans la figure). Ces lois d'échelles généralisées permettent donc de prédire comment choisir à la fois la composition chimique et la structure de surface d’un catalyseur afin de le rendre optimal pour une réaction donnée.
Ce travail a été développé et financé dans le cadre du contrat Européen PUMA MIND (Grant n°303419; Call FCH-JU-2011-1; Code SP1-JTI-FCH.2011.1.3).
Référence : Federico Calle-Vallejo, David Loffreda, Marc T. M. Koper and Philippe Sautet, Introducing structural sensitivity into scaling relations between adsorption energies by means of coordination numbers, Nature Chemistry 6 avril 2015, DOI : 10.1038/NCHEM.2226
Disciplines
Mots clés